MCSE-Free CRM Performance and Safety Assessment
David C. Norris
4/25/2021
Source:vignettes/MCSE-Free.Rmd
MCSE-Free.Rmd“In most trials, and in particular in cancer chemotherapy trials, it is rare that we have no idea at all about the dose-response relationship. Such information is implicitly used in the choice of dose levels available for use in the trial, and part of our approach here is to make an attempt to quantify such information.”1
“It is important to see to what extent the largely arbitrary specification of these features influences the operational nature of the method, as well as the conclusions reached. Experimenters prefer methods which are relatively insensitive to prior specification as well as the form of the working model.”2
“Most clinicians do not feel sufficiently confident in their initial toxicity probability estimates to start above the lowest dose (often chosen to be 10% of rodents’ LD\(_{10}\)).”3
“The main challenge when using the CRM is model calibration.”4
Background
Since its inception 3 decades ago (O’Quigley, Pepe, and Fisher 1990), the CRM has retreated from the sincere Bayesianism of its original aim to manifest clinically relevant prior information. The early retreat was quite swift, in fact. Section 2.4 of O’Quigley, Pepe, and Fisher (1990), titled ‘Establishing Dose Levels’, suggested a substantial collaboration between modelers and clinical investigators. Yet within a few years, Chevret (1993) already accepted “that \(k\) distinct doses are chosen for experimentation, defined by the investigator through an implicit idea of the dose-toxicity relationship, however imprecise,” and demoted the CRM’s prior dose-toxicity curve to the status of a (multidimensional) tuning parameter. The retreat continued apace with Goodman, Zahurak, and Piantadosi (1995) giving voice to the deep skepticism with which clinical investigators were already viewing the CRM. Indeed, a key “practical improvement” for which that paper is still cited, was its rejection of the prior curve’s pretense to inform the trial starting dose.
By twenty years later, biostatisticians had completed their retreat, with the prior toxicity curve being variously rebranded to avoid using the word ‘prior,’5 and computer-based model tuning activities having become “the main challenge when using the CRM” (Lee and Cheung 2011). Biostatisticians remain to this day intent on problems arising out of CRM calibration practice, as demonstrated in the recent contribution by Braun (2020), who develops a mean-field approximation that greatly accelerates CRM performance characterization and calibration. Applied to a previously-completed trial, this approximation technique reduces certain calibration steps from 6.3 hours to 18 seconds (1260\(\times\)) and from 2 days to 2 minutes (\(\approx 1400 \times\)).
Here I revisit Braun’s application from the perspective of complete path enumeration (CPE), which package precautionary now implements for the CRM.
Performance characterization for a given CRM model
Prerequisite to any CRM prior calibration procedure aiming to optimize selected performance characteristics, is the ability to characterize performance for any given CRM model with fixed prior parameters.6 So we begin by repeating Braun’s characterization of his calibrated model.7 The calibrated model is as follows:
skel_0 <- c(0.03, 0.11, 0.25, 0.42, 0.58, 0.71)
calmod <- Crm$new(skeleton = skel_0,
scale = 0.85, # aka 'sigma'
target = 0.25)Braun’s initial characterization is of trials of fixed enrollment \(N=30\), with a cohort size of 2.8 Trials of this size readily yield to CPE on desktop hardware:
d1_maxn <- 5
cum_maxn <- 10
system.time({
calmod$
no_skip_esc(TRUE)$ # compare Braun's 'restrict = T'
no_skip_deesc(FALSE)$
stop_func(function(x) {
enrolled <- tabulate(x$level, nbins = length(x$prior))
x$stop <- enrolled[1] >= d1_maxn || max(enrolled) >= cum_maxn
x
})$trace_paths(root_dose = 1,
cohort_sizes = rep(2, 15) # ==> N=30
)
T <- calmod$path_array()
})## T (22041×15×6) constructed in 0.870 sec
## T condensed in 0.811 sec## user system elapsed
## 5.365 0.165 4.732The 3-dimensional array T[j,c,d] has the same structure and function as in Norris (2020):
dim(T)## [1] 22041 5 6Along each of the 22041 paths enumerated, toxicities have been tabulated separately for as many as 5 distinct cohorts enrolled at each of 6 doses.
Again as in Norris (2020), we may …
Compute \(\mathrm{U}\) and \(\mathrm{Y}\)
Y <- apply(T, MARGIN = c(1,3), FUN = sum, na.rm = TRUE)
Z <- apply(2-T, MARGIN = c(1,3), FUN = sum, na.rm = TRUE)
U <- cbind(Y, Z)
dim(U)## [1] 22041 12Express \(\mathbf{\pi}\) as a function of dose-wise DLT probabilities
log_pi <- function(prob.DLT) {
log_p <- log(prob.DLT)
log_q <- log(1 - prob.DLT)
b + U %*% c(log_p, log_q)
}Note in particular that \(\mathbf{\pi}(\mathbf{p})\) is a function of the DLT probabilities at the design’s prespecified doses, and so is defined on the simplex \(0 < p_1 < p_2 < ... < p_D < 1\). To demonstrate that the constraint \(\sum_j \pi^j \equiv 1\) holds over this domain, let us check the identity at a randomly chosen point:9
p <- sort(runif(6)) # randomly select a DLT probability vector
sum(exp(log_pi(p))) # check path probabilities sum to 1## [1] 1Together with this ability to compute the path probabilities \(\mathbf{\pi}(\mathbf{p})\) under any scenario \(\mathbf{p}\), the complete frequentist characterization by \(\mathrm{T}\) of all path outcomes enables us immediately to obtain any of the common performance characteristics of interest:10
Probability of Correct Selection (PCS)
Whereas the array \(T_{c,d}^j\) contains clinical events (enrollments, toxicities) along each path, the final dose recommendations are retained as the rightmost non-NA value in each row of calmod’s path matrix.11 The path_rx() method of the Crm class returns this:
xtabs(~calmod$path_rx())## calmod$path_rx()
## 1 2 3 4 5 6
## 6178 9266 4864 1314 341 78These dose recommendations of course must be weighted by the path probabilities of some chosen dose-toxicity scenario. For sake of an easy demonstration, let’s take our scenario from the skeleton itself:
pi_skel <- calmod$path_probs(calmod$skeleton())
xtabs(pi_skel ~ calmod$path_rx())## calmod$path_rx()
## 1 2 3 4 5 6
## 0.010420 0.248309 0.560418 0.172114 0.008630 0.000109According to our skeleton, dose 3 is ‘the’ MTD; so PCS is 0.56 in this scenario.
Fraction Assigned to MTD
The array \(T_{c,d}^j\) lets us count patients assigned to ‘the’ MTD:
path_cohorts <- apply(!is.na(T), MARGIN=1, FUN=sum, na.rm=TRUE)
path_MTDcohs <- apply(!is.na(T[,,3]), MARGIN=1, FUN=sum, na.rm=TRUE)
sum(pi_skel * path_MTDcohs / path_cohorts)## [1] 0.3895Fraction ‘Overdosed’
A similar calculation enables us to calculate the fraction assigned to doses exceeding ‘the’ MTD:
path_ODcohs <- apply(!is.na(T[,,4:6]), MARGIN=1, FUN=sum, na.rm=TRUE)
sum(pi_skel * path_ODcohs / path_cohorts)## [1] 0.1971Furthermore, we can ask what fraction of thus-‘overdosed’ trial participants may be expected to exceed their own MTD\(_i\)’s:12
path_DLTs4_6 <- apply(T[,,4:6], MARGIN=1, FUN=sum, na.rm=TRUE)
sum(pi_skel * path_DLTs4_6) / sum(pi_skel * 2*path_ODcohs)## [1] 0.442Extensions to scenario ensembles
As the examples above illustrate, once a fixed trial design has been completely path-enumerated, its performance characteristics are available through fast array operations. The generalization of single-scenario performance evaluations to ensemble-average evaluations should prove straightforward.
Extending to larger trials
saved <- options(mc.cores = 6)
kraken <- data.frame(C = 11:25, J = NA_integer_, elapsed = NA_real_)
for (i in 1:nrow(kraken)) {
C <- kraken$C[i]
calmod$skeleton(calmod$skeleton()) # reset skeleton to clear cache for honest timing
time <- system.time(
calmod$trace_paths(1, rep(2,C), impl='rusti', unroll=8))
kraken$elapsed[i] <- time['elapsed']
kraken$J[i] <- dim(calmod$path_matrix())[1]
print(kraken)
}
options(saved)| C | J | elapsed |
|---|---|---|
| 11 | 5001 | 2.06 |
| 12 | 7839 | 2.94 |
| 13 | 11571 | 3.96 |
| 14 | 16125 | 5.05 |
| 15 | 22041 | 7.17 |
| 16 | 28485 | 8.94 |
| 17 | 35589 | 11.02 |
| 18 | 43371 | 13.41 |
| 19 | 48255 | 14.70 |
| 20 | 50379 | 14.91 |
| 21 | 52011 | 15.30 |
| 22 | 53085 | 15.42 |
| 23 | 53205 | 15.48 |
| 24 | 53205 | 15.52 |
| 25 | 53205 | 15.81 |
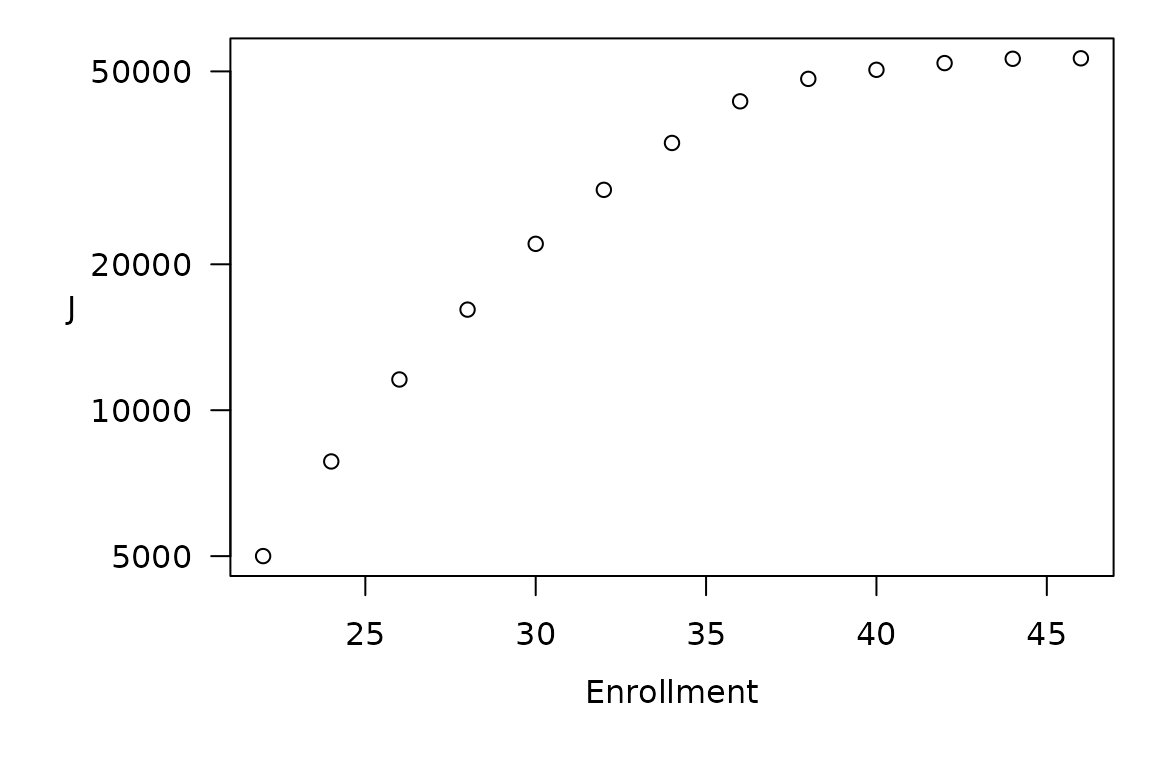
Number of paths J vs enrollment for the CRM trial at hand. Note that all paths hit stopping criteria no later than at N = 46 (23 cohorts of 2), so that what begins as exponential growth (the linear trend for N < 30 in this semi-log plot) rapidly succumbs to this hard upper bound.
Reassuringly, the (perhaps typical) stopping criteria in this trial impose a finite upper bound on CPE size stringent enough to keep the space and time complexity of CPE feasible. Even without such planned stopping rules, as a practical matter, once a dose-finding trial has enrolled more than 30 participants, it ought to have yielded enough information to warrant revisiting the original design. This limits the reasonable ‘time between overhauls’13 for a fixed CRM trial design. Thus, CPE-based performance characterization may prove entirely feasible for most practical applications of the standard CRM model.14
Calibration of CRM priors
The foregoing analysis shows that CPE-based performance characterization for a given CRM model of typical size may require on the order of 15 seconds on quite modest hardware. To calibrate a CRM model via CPE, however, may incur this cost repeatedly—perhaps hundreds of times—during what will generally amount to the derivative-free optimization of a discontinuous objective function over the prior-parameter space. For the example at hand, the Nelder-Mead method finds a PCS-optimal skeleton in just 25 minutes:
scenario <- skel_0 # for a demo scenario, we use our original skeleton
pcs <- function(s, unroll=6) { # unroll=6 yields quickest PCS for C=15 case
if (any(diff(s) <= 0) || s[1] <= 0 || s[length(s)] >= 1)
return(NA) # out-of-bounds
calmod$skeleton(s)
calmod$trace_paths(1, rep(2,15), impl='rusti', unroll=unroll)
pi_0 <- calmod$path_probs(scenario)
correct <- calmod$path_rx() == 3
sum(pi_0[correct])
}
optim(calmod$skeleton(), pcs, method="Nelder-Mead",
control = list(fnscale = -1, # NEGative ==> MAXimize PCS
reltol = 0.001, trace = 2))Exiting from Nelder Mead minimizer
135 function evaluations used
$par
[1] 0.03040 0.08371 0.23867 0.43310 0.59292 0.72230
$value
[1] 0.5931
$counts
function gradient
135 NA
$convergence
[1] 0
$message
NULLMiscellaneous helps to optimization
Cheung and Chappell (2002) develop an asymptotic assessment capable of rejecting certain skeletons as intrinsically unreasonable because they confer inadequate sensitivity on the CRM. This criterion applies irrespective of dose-toxicity scenario, and so might usefully restrict our skeleton search during calibration by Nelder-Mead or similar direct-search method. The dimensionality of this search might also be addressed directly, by parametrizing the skeleton in 2 or 3 suitable basis dimensions. But such exquisite efforts risk taking this type of calibration exercise too seriously.
On calibrating ‘priors’
In everyday usage, calibration suggests an exacting procedure, in which typically a measuring instrument is adjusted to accord with some objective standard or criterion. In statistics, however, the term may not carry quite this connotation. Certainly, Grieve (2016) discusses the calibration of Bayesian trial designs generally in much the same sense as our ‘CRM calibration’ here, with the criterion being provided by whatever frequentist properties are deemed desirable in each given application.
Whatever we call this chiropractic of the CRM skeleton, the very act of treating the CRM’s prior dose-toxicity probabilities as free parameters undermines the presumption that model-basedness per se necessarily confers special qualities upon dose-finding methods. But now that even the CRM—the very type specimen of model-based dose finding—yields to complete path enumeration, ‘model-based’ has become a distinction without a difference.
References
Braun, Thomas M. 2020. “A Simulation‐free Approach to Assessing the Performance of the Continual Reassessment Method.” Statistics in Medicine, September, sim.8746. https://doi.org/10.1002/sim.8746.
Cheung, Ying Kuen, and Rick Chappell. 2002. “A Simple Technique to Evaluate Model Sensitivity in the Continual Reassessment Method.” Biometrics 58 (3): 671–74. https://doi.org/10.1111/j.0006-341x.2002.00671.x.
Chevret, S. 1993. “The Continual Reassessment Method in Cancer Phase I Clinical Trials: A Simulation Study.” Statistics in Medicine 12 (12): 1093–1108. https://doi.org/10.1002/sim.4780121201.
Goodman, S. N., M. L. Zahurak, and S. Piantadosi. 1995. “Some Practical Improvements in the Continual Reassessment Method for Phase I Studies.” Statistics in Medicine 14 (11): 1149–61.
Grieve, Andrew P. 2016. “Idle Thoughts of a ‘Well-Calibrated’ Bayesian in Clinical Drug Development.” Pharmaceutical Statistics 15 (2): 96–108. https://doi.org/10.1002/pst.1736.
Lee, Shing M., and Ying Kuen Cheung. 2009. “Model Calibration in the Continual Reassessment Method.” Clinical Trials (London, England) 6 (3): 227–38. https://doi.org/10.1177/1740774509105076.
———. 2011. “Calibration of Prior Variance in the Bayesian Continual Reassessment Method.” Statistics in Medicine 30 (17): 2081–9. https://doi.org/10.1002/sim.4139.
Norris, David C. 2020. “What Were They Thinking? Pharmacologic Priors Implicit in a Choice of 3+3 Dose-Escalation Design.” arXiv:2012.05301 [stat.ME], December. https://arxiv.org/abs/2012.05301.
O’Quigley, J., M. Pepe, and L. Fisher. 1990. “Continual Reassessment Method: A Practical Design for Phase 1 Clinical Trials in Cancer.” Biometrics 46 (1): 33–48.
Wages, Nolan A, Mark R Conaway, and John O’Quigley. 2011. “Dose-Finding Design for Multi-Drug Combinations.” Clinical Trials: Journal of the Society for Clinical Trials 8 (4): 380–89. https://doi.org/10.1177/1740774511408748.
O’Quigley, Pepe, and Fisher (1990)↩︎
Chevret (1993)↩︎
Goodman, Zahurak, and Piantadosi (1995)↩︎
Lee and Cheung (2011)↩︎
For example, Lee and Cheung (2009) exploit a technicality of the CRM to identify this prior curve with “the selection of ‘dose levels’”, while Wages, Conaway, and O’Quigley (2011) apparently introduce the now-popular ‘skeleton’.↩︎
This is just the observation that we must be able to calculate an objective function in order to optimize it.↩︎
This starts at the bottom of page 8 in Braun (2020).↩︎
The cohort size is not mentioned in the text, but see the example at the bottom of Appendix 1, where Braun sets
cohort_size <- 2.↩︎But note this constraint holds trivially, simply by the construction of \(\mathrm{U}\) from complementary left and right halves, \(\mathrm{Y}\) and \(\mathrm{Z}\).↩︎
I suspend judgment as to whether we should do this, and indeed whether these performance characteristics make any sense at all.↩︎
This matrix retains the columnar layout—although not the row degeneracy—of the dose transition pathways (DTP) tables of package
dtpcrm↩︎That is, to experience a DLT. Note that this latter fraction constitutes a patient-centered notion of ‘overdose’, in contrast to the traditionally dose-centered notion.↩︎
This perhaps does not apply to applications such as TITE CRM, where participants are modeled non-exchangeably. CPE for TITE CRM remains an open problem.↩︎